
SNAPR: A Bioinformatics Pipeline for Efficient and Accurate RNA-Seq Alignment and Analysis
By Andrew T. Magis, Cory C. Funk, and Nathan D. Price
Published August 28, 2015.
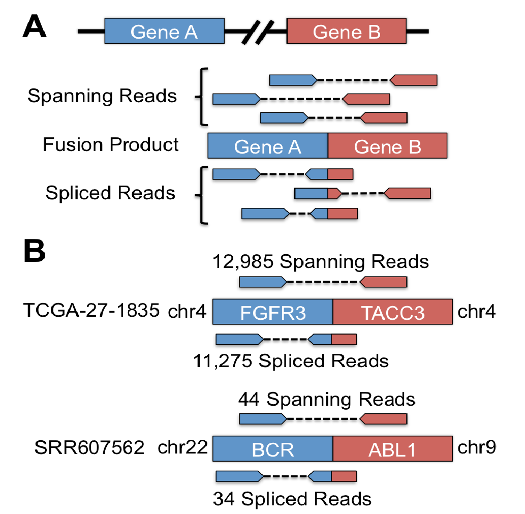
The process of converting raw RNA sequencing (RNA-seq) data to interpretable results can be circuitous and time-consuming, requiring multiple steps. We present an RNA-seq mapping algorithm that streamlines this process. Our algorithm utilizes a hash table approach to leverage the availability and the power of high memory machines. SNAPR, which can be run on a single library or thousands of libraries, can take compressed or uncompressed FASTQ and BAM files, and output a sorted BAM file, individual read counts, and gene fusions, and can identify exogenous RNA species in a single step. SNAPR also does native Phred score filtering of reads. SNAPR is also well suited for future sequencing platforms that generate longer reads. We show how we can analyze data from hundreds of TCGA samples in a matter of hours while identifying gene fusions and viral events at the same time. With the reference genome and transcriptome undergoing periodic updates and the need for uniform parameters when integrating multiple data sets, there is great need for a streamlined process for RNA-seq analysis. We demonstrate how SNAPR does this efficiently and accurately.
 Interim Editor-in-Chief
Interim Editor-in-Chief  Senior Editor for Personalized Medicine
Senior Editor for Personalized Medicine  Senior Editor for Pharmaceutical Engineering
Senior Editor for Pharmaceutical Engineering  Senior Editor for Synthetic Biology
Senior Editor for Synthetic Biology  Senior Editor for Systems Biology
Senior Editor for Systems Biology