Genome Sequencing To Identify Driver Pathways in Cancer
By Fabio Vandin, Eli Upfal, and Benjamin J. Raphael
NOTE: This is an overview of an article which appeared in the March 2012 issue of the IEEE Computer magazine.
Click here to read the entire article.
Cancer is a disease driven by mutations in an individual’s DNA sequence, or genome, that accumulate during the person’s lifetime. These mutations arise during DNA replication, which occurs as cells grow and divide into two daughter cells. They range from single “character” substitutions (the nucleotides A, C, T, and G of DNA) to structural variants that duplicate, delete, or rearrange larger genome segments. Single-nucleotide substitutions occur at a rate of approximately 10-9, so that on average each daughter cell contains around six somatic mutations. Most are benign, or inconsequential for the organism. However, in certain circumstances, dangerous mutations can accumulate in a collection of cells and lead to cancer.
In some types of leukemia, for example, chromosomes 9 and 22 undergo a translocation that swaps DNA between these chromosomes. Unfortunately, finding other important large-scale rearrangements has been a challenge. Many cancer cells contain dozens of chromosomal abnormalities, and these differ among individuals with the same type of cancer. A natural question is whether some or all of these rearrangements contribute to cancer or are merely random occurrences.
The emergence of DNA sequencing has enabled biologists to measure single-nucleotide mutations with increasing speed and accuracy. These studies showed that the “typical” cancer genome might have hundreds to thousands of somatic mutations of different types. However, most of the somatic mutations in a cancer cell are benign passenger mutations. A much smaller fraction of driver mutations are important for cancer development, with current estimates ranging from 10 to 20 driver mutations per tumor. Because cancer cells have a large variety of relatively rare mutations, genome-wide studies for identifying cancer driver mutations require sequencing numerous patients. This task became feasible in the past five years with the development of next-generation sequencing technologies.
A key question for such projects is how to use the resulting DNA sequence to understand the mutations that cause specific properties of cancer cells.
This article points out computational challenges in applying next-generation DNA sequencing to cancer genomes. The first is how to derive catalogs of mutations in a genome from the data generated by a DNA sequencing machine. Although these machines produce a remarkable number of DNA sequences, these sequences are only reads (about 30 to 1,000 nucleotides), not full-length genomes. Obtaining the catalog of somatic mutations from such short sequences requires algorithmic techniques, an active area of investigation in recent years. Assuming we have obtained a list of all somatic mutations in the cancer genome, the second challenge is to distinguish the driver mutations from the random passenger mutations. One way to predict candidate driver mutations is to examine the somatic mutations measured in a large population of cancer patients and identify recurrent mutations that occur more frequently than expected by chance, or, alternatively, recurrently mutated genes, which are genes that are mutated more frequently than expected. The article describes a probabilistic model for identifying cancer mutations, based on a coin-flipping experiment. This model can help determine whether the number of mutated patients is significantly higher than expected. If so, the gene is a candidate driver gene in cancer. This test (with a more detailed model for the passenger mutation rate) is essentially current practice in cancer genome projects.
There are several reasons that the current practice, fails to identify many driver mutations. One is that driver mutations target groups of genes, or pathways. Genes do not act in isolation, but rather interact with other genes (and the proteins these genes produce) in complex signaling and regulatory networks.
Appropriate algorithms can significantly reduce the number of combinations of possible driver mutations that need to be tested in expensive and time-consuming wet lab experiments. These algorithms need to be time and space efficient to handle massive datasets, and they must rely on rigorous statistical methods to reduce the number of candidate driver mutations (false positives) while also not eliminating true driver mutations from consideration (false negatives).
Both of the algorithms that the authors designed to find groups of genes that are functionally redundant for the development of cancer generalize the single-gene test that is commonly used to identify driver genes by their recurrence in many cancer patients. Moreover, neither algorithm restricts groups of genes to those already known to be involved in cancer, thus allowing the discovery of novel combinations of mutations. The two algorithms presented in this article, along with results of their application. address this.
The first algorithm, named ‘HotNet’ (See Figure 1), considers the mutation data for many patients to identify ‘hot’ subnetworks of mutated genes. The HotNet algorithm relies on prior knowledge of the interactions between genes, represented as a graph, to restrict the search space of possible combinations.
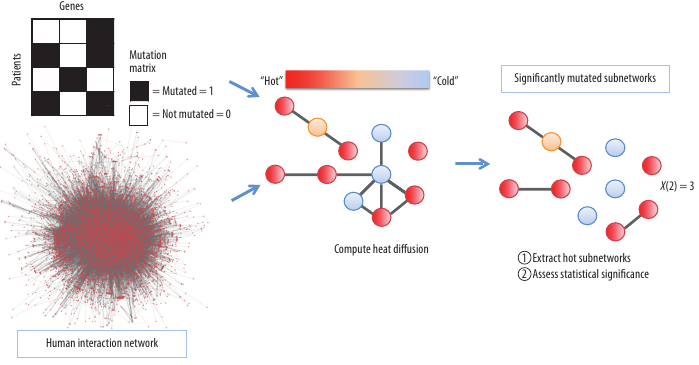
Figure 1. The HotNet algorithm combines mutation data and protein-protein interaction network information to find hot subnetworks, or clusters of interacting genes, that are mutated in a significant number of cancer patients. On each gene, HotNet places a source of heat proportional to the number of mutations on the gene. The heat diffuses on the network for a fixed time, revealing the hot subnetworks. Finally, a statistical test assesses the significance of the list of observed subnetworks.
![]()
The second, named ‘Dendrix’ (Figure 2), identifies sets of genes involved in cancer signalling pathways. The Dendrix algorithm exploits some combinatorial properties of the patterns of mutations that are expected for driver pathways.
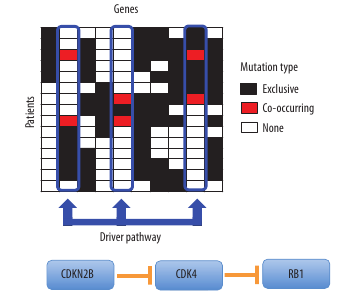
Figure 2. The De novo Driver Exclusivity (Dendrix) algorithm uses a Markov chain Monte Carlo method to sample submatrices of the mutation matrix with high coverage and exclusivity. In this case, Dendrix identifies a set of three genes – CDKN2B, CDK4, and RB1 – in brain cancer data corresponding to a known important pathway.
The article notes that more research is required to develop better algorithms for the identification of driver genes and driver pathways, and to use the resulting information to improve cancer treatments. While the article focused on mutation data, a wealth of other types of genomic and epigenomic data – on gene expression, DNA methylation, and so on – can be combined to make more accurate predictions. The Cancer Genome Atlas and other similar projects are collecting multiple data types on the same patients that can be used for such research. Finally, identifying the driver mutations and pathways is only a first step toward understanding how these mutations affect a particular patient’s prognosis and treatment.
ABOUT THE AUTHORS
Fabio Vandin is a research assistant professor in the Department of Computer Science at Brown University. His research interests include the design of algorithms and applications for cancer genomics, structural proteomics, biological sequence analysis, and database mining. Vandin received a PhD in information engineering from the University of Padua, Italy. He is a member of the International Society for Computational Biology and the European Association for Theoretical Computer Science. Contact him at vandinfa@cs.brown.edu.
Eli Upfal is a professor in the Department of Computer Science at Brown University. His research focuses on the design and probabilistic analysis of algorithms, and on statistical methods in computation. Upfal received a PhD in computer science from the Hebrew University of Jerusalem, Israel. He is a Fellow of IEEE and ACM. Contact him at eli@cs.brown.edu.
Benjamin J. Raphael is an associate professor in the Department of Computer Science at Brown University. His research focuses on the design of combinatorial and statistical algorithms for the interpretation of genomes. Raphael received a PhD in mathematics from the University of California, San Diego, and completed postdoctoral training in bioinformatics and computer science at UCSD. Contact him at braphael@cs.brown.edu.
